


【化工進展】北京低碳清潔能源研究院 | 張琪,門卓武,呂毅軍,等:合成氣制高級醇Co基催化劑研究進展
合成氣制高級醇Co基催化劑研究進展
張琪,王濤,張雪冰,李為真,馮波,蔣智慧,呂毅軍,門卓武
北京低碳清潔能源研究院,北京 102209
引用本文
張琪, 王濤, 張雪冰, 等. 合成氣制高級醇Co基催化劑研究進展[J]. 化工進展, 2025, 44(2): 773-787.
?
DOI:10.16085/j.issn.1000-6613.2024-0129

摘要
合成氣高選擇性制取高級醇對推動煤炭清潔高效利用具有重要意義。Co基催化劑的雙活性位點對高級醇合成反應具有獨特優(yōu)勢。CoCu催化劑因材料成本低、高級醇產量高,成為近年來的研究熱點,但其雙功能活性位點作用機制尚不清晰,基礎理論研究仍待完善。本文概述了Co基催化劑的最新研究進展,重點綜述了Co基催化劑在合成高級醇反應中活性位點的性質和反應機理,厘清了Cu/Co比例、CoCu分布均勻性對催化劑性能的影響關系。梳理了堿金屬、Zr、Ga等助劑對催化劑構效關系的影響,及其對改善碳鏈生長、抑制甲烷化、防止碳沉積、提高穩(wěn)定性等作用。系統(tǒng)論述了具備獨特層狀結構的LDHs,及CNTs、AC等眾多碳基材料作載體對Co基催化劑性能的影響。通過深度剖析合成氣制高級醇機理,針對性開發(fā)新型催化劑結構及載體材料,可進一步提高Co基催化劑性能,實現更高效、環(huán)保和可持續(xù)的高級醇合成,為實現商業(yè)化夯實基礎。
富煤貧油少氣是我國一次能源結構的特點,加強煤炭清潔高效利用可推動現代煤化工高端化、多元化、低碳化發(fā)展。合成氣高效利用是煤化工行業(yè)提高能源效率的關鍵,合成氣下游產品含氧化合物可拉動高分子材料、有機及醫(yī)藥化學品產業(yè)鏈。以合成氣高選擇性地合成乙醇等C2+含氧化合物一直是碳一化學領域中的重要課題,其中C2+醇可稱為高級醇(HA),廣泛用作潛在燃料添加劑、氫載體、潤滑劑及醫(yī)藥中間體,是化學和聚合物行業(yè)合成商品和特殊產品的重要原料。生產HA可通過糖發(fā)酵(乙醇和異丁醇)、石油衍生的烯烴水合反應(較重的醇)、合成氣直接合成高級醇(HAS)等途徑。其中HAS原料可取自非常規(guī)天然氣、生物質甚至CO2,有效緩解能源危機和環(huán)境污染問題,具有明顯可持續(xù)性優(yōu)勢。
HAS反應研究始于1930年代,油價變化、新的天然氣及合成氣來源等因素促進著研發(fā)興趣,尤其在新型催化劑配方及改性等方面。催化劑活性低、穩(wěn)定性差、壽命短及目標產物選擇性低是制約HAS技術大規(guī)模工業(yè)化的瓶頸。用于HAS的催化劑種類繁多,通常可分為銠(Rh)基催化劑、鉬(Mo)基催化劑、改性費托合成(FTS)催化劑和改性甲醇合成(MS)催化劑四類。根據提供活性中心的來源不同,其也可分為單金屬催化劑、雙金屬催化劑和三金屬/多金屬催化劑,及特定類別的HAS催化劑。HAS結合了FTS和MS反應,催化劑應具備雙位點雙功能,C—C耦合和CO加氫兩種活性位點,一種解離CO并形成表面烷基物種,另一種催化非解離CO吸附以插入CO和形成醇。
Co基催化劑的雙活性位點對HAS具有獨特優(yōu)勢,加入第二種金屬有助于耦合CO分子吸附中心與解離吸附中心,CoCu、CoMn等組合是近年來研究的熱點。因材料成本低、HA產量高,CoCu基催化劑活性位點性質被廣泛關注。Co催化CO解離,Cu存在爭議,作用為CO吸附和插入,亦或是調節(jié)Co0與Con+比例而影響催化性能。Co和Cu高分散且緊密接觸是提高HA選擇性的必要條件,然而Cu表面能比Co高,催化劑還原過程中易聚集在表面,難以控制Co和Cu分布均勻,存在Co和Cu相間配位低和穩(wěn)定性差等問題。調節(jié)Cu/Co配比、將活性金屬和促進劑緊密混合到具有規(guī)定結構和孔隙率的基質中等,是提高催化劑活性和選擇性的有效手段。同時應關注反應過程中金屬團聚或析出導致的結構穩(wěn)定性、烷烴和CO2選擇性變化,確保在合成氣高轉化率的基礎上提高HA選擇性。
01
機理
HAS反應涉及CO和H2解離、CO氫化、CO插入及烷基部分的形成和偶聯(lián),催化劑應具有CO解離吸附和非解離吸附的雙中心。非解離吸附的CO直接加氫傾向于生成甲醇,C—C耦合中間體直接加氫則更易生成烷烴和烯烴,僅在CO插入和C—C耦合動力學匹配時方可有效提高HA選擇性。雙功能中心應分布均勻,方可充分發(fā)揮CO解離吸附和非解離吸附活性位點間的協(xié)同作用。
從CO活化到形成乙醇或HA的最佳路徑取決于金屬特性和促進劑等。反應機理重點涉及CO活化、CHx形成、CO插入和逐步氫化為醇,圖1為CO活化為CHx途徑及CO插入機制。吸附的CO直接解離或在H輔助下解離,直接解離產生C原子隨后被氫化為CHx,而H輔助解離產生CHO(優(yōu)選)或COH作為中間體,再被繼續(xù)氫化解離為CHx。CO/CHO插入CHx是生成C2含氧化合物的關鍵步驟,產生CHxCO或CHxCHO,進一步加氫生成乙醇。但具體加氫順序仍待解析,中間體可能是CH3CH2O或CH3CHOH。
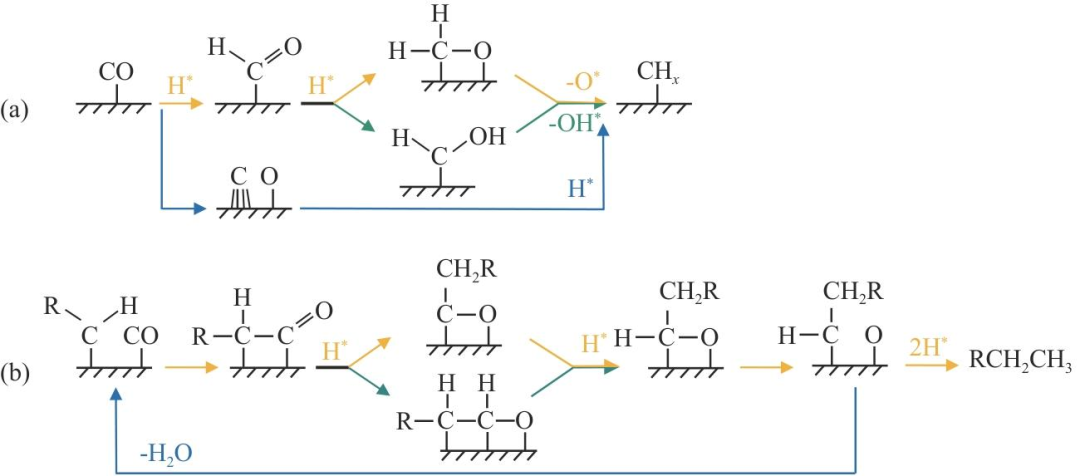
CO活化為CHx途徑及CO插入機制
Co是最常用的活性金屬之一,Co2C可有效地將合成氣轉化為HA和高碳烴(HC),但Co2C同時也有甲烷選擇性高、易失活的缺點,其性質和形成條件尚不明確。Pei等認為金屬Co實現CO解離吸附,是鏈生長的原因,而碳化物為CO分子吸附提供了位點,CO插入優(yōu)先發(fā)生在Co-Co2C界面。Lebarbier等對比Co2C和Co上的CO解離、CH氫化和CO插入,發(fā)現能量上CO解離和碳鏈生長(CHx+CHy)在金屬Co上比在Co2C上更有利,CH是最穩(wěn)定的中間體。結合密度泛函理論(DFT)和微觀動力學模擬,Inderwildi等認為在平坦的Co(0001)表面上CO并非直接解離,而是在H輔助下進行CO解離,CHO和CH2O是中間體[圖1(a)黃色],而Ojeda等認為中間體是CHO和CHOH[圖1(a)綠色]。Zhuo等與Inderwildi等觀點一致,認為從CO形成CHO和CH2O有效降低了C—O鍵的解離勢壘,從220kJ/mol降低到90kJ/mol,并提出CO插入RCH表面先形成RCHCO,繼續(xù)添加H產生RCH2CHO醛,C—O斷裂后釋放RCH2CH3[圖1(b)]。Zhao等研究發(fā)現HAS反應中表面條件使部分金屬Co先轉變?yōu)镃o2C,再形成Co@Co2C納米顆粒(NPs),小于6nm的殘余小Co0團簇分布在Co2C NPs(約20nm)表面,使金屬Co和Co2C相之間發(fā)生協(xié)同效應。
Co基催化劑加入Cu產生協(xié)同催化可促進HA生成,但CoCu基催化劑活性位點性質存在爭議,另一種觀點認為Cu僅改變Co還原性、調變Co0/Con+摩爾比。為闡明Cu的作用,蘇俊杰等認為煅燒后Cu進入Co3O4晶格形成混合尖晶石結構,Cux2+Co1-x2+Co23+O4尖晶石是由低Cu濃度下以四面體Co2+陽離子取代CuCo2O4中Cu2+離子而產生的。還原后呈類合金CuCo雙金屬相并存在偏析,CuCo雙金屬相表面富集金屬Cu并覆蓋大部分Co表面,形成非連續(xù)Co活性位,表面僅剩部分低配位低聚態(tài)Co位。基于動力學研究認為Cu的作用是通過削弱CO/HCO解離來減少CHx的形成,并控制可以抑制CHx插入的表面Co的整體尺寸。Smith等通過原位XRD、XANES和紅外漫反射光譜(DRIFTS)研究Co/SiO2、Cu/SiO2和CoCu/SiO2的還原和CO吸附行為。發(fā)現在Co/SiO2中Co0和Con+兩個位點分別負責CO的解離吸附和分子吸附,見圖2。Co的存在不會改變Cu位點上的CO吸附頻率和結合強度,因此認為在后兩個樣品間HA選擇性的差異并非Cu的作用。為深入研究Cu和Co結構域間協(xié)同催化作用,Lyu等在Co納米片上沉積Cu納米團簇,基于HRTEM和催化結果提出HA的生成源于Cu(111)和Co(100)表面之間的界面,然而高達93%的HC選擇性引發(fā)了對研究結論相關性的質疑。
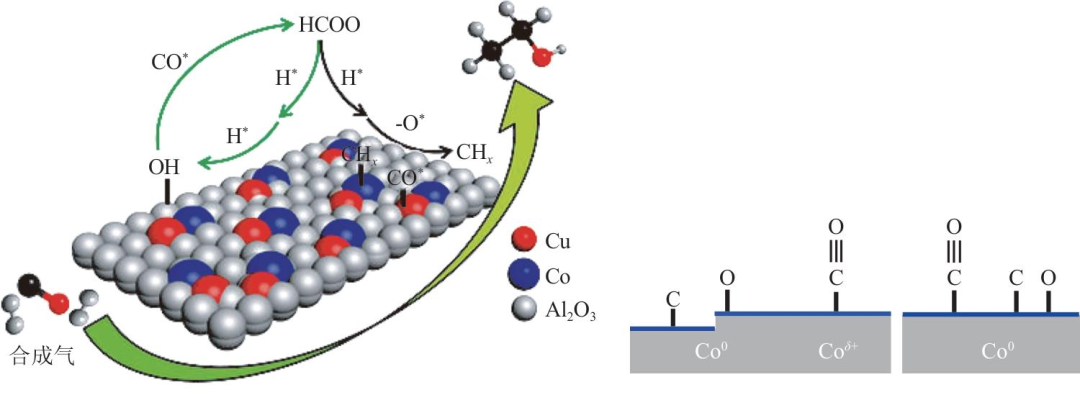
CoCu雙金屬協(xié)同作用與單金屬Co作用機理對比
碳鏈生長過程可通過DFT計算及kMC方法模擬,Yu等認為CH2和CH3是實現鏈生長的關鍵中間體,在CoCu(100)上很容易形成。Co可以促進CH2O的C==O鍵斷裂形成CH2,抑制CH3O的形成,改變CoCu(100)上的反應途徑,從而提高C2+醇的選擇性。Wang等認為在Co(0001)上CO插入CH3步驟比其他CHx偶聯(lián)活化能更高,Cu和Co結構域間邊界通過減少該反應的勢壘來促進鏈生長。在圖3各種表面中,雙金屬Cu/Co-Ⅲ表面的活化能最低,具有中等吸附強度,是締合反應適宜的表面,Cu/Co-Ⅱ的活化能略高。CAO等研究在CuCo緊密堆積(111)和階梯狀(211)表面上形成甲烷、甲醇和乙醇的過程。發(fā)現CuCo合金表面上可以得到促進C-C偶聯(lián)所需的CO和CHx覆蓋率,并且選擇性對合金結構非常敏感。部分反應條件下CuCo(211)表面對乙醇具有高活性和選擇性,歸因于C—O解離屏障低以及CHx-CO偶聯(lián)速率高,而CuCo(111)表面則對甲醇具有選擇性。
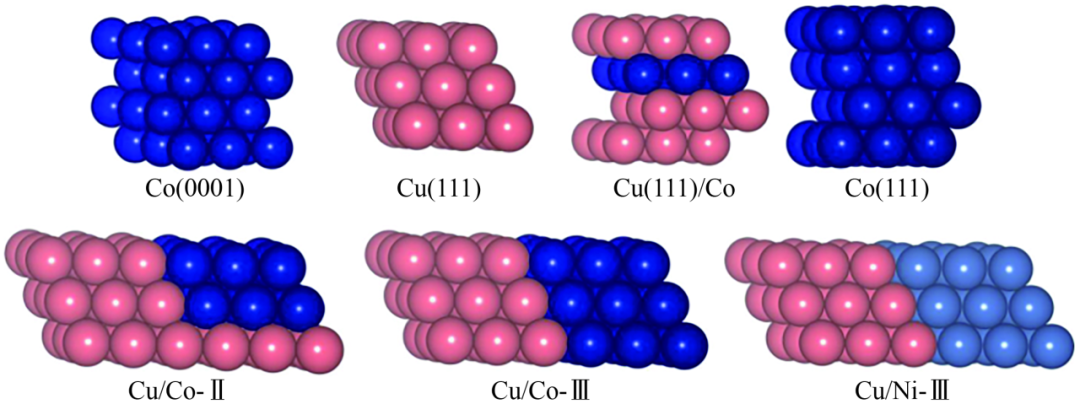
雙金屬Cu/Co表面結構
02
單金屬Co催化劑
Co基催化劑碳鏈生長能力強且對水煤氣變換反應不敏感,是HAS領域最有前途的催化劑體系之一。單金屬Co催化劑中同時存在Con+和Co0,因此單獨使用Co即可產生HA,但非解離吸附CO的活性中心少,產物醇選擇性低。
Coδ+被認為與Cu具有類似作用,可作為CO非解離吸附及插入的活性位點,與Co0形成Coδ+/Co0活性對,提高HA選擇性。Ning等通過Co-CoOx/MAO催化劑研究Co0和Coδ+雙活性中心,認為Co-CoOx/MAO優(yōu)異的C-C偶聯(lián)性能歸因于Co0位點對CO2的有效吸附和活化,從而產生C1中間體,而引入Coδ+可有效降低關鍵CHCH*中間體的能壘。Song等研究CoNPs與LaAl1-xCoxO3相互作用,發(fā)現還原后的Co-LaAlO3/ZrO2催化劑中Co呈Coδ+/Co0活性對狀態(tài)。Co NPs與LaAlO3相互作用使兩者界面處形成Coδ+,并且可通過Co NPs大小調節(jié)Coδ+/Co0比例。Coδ+/Co0活性對中Coδ+可締合吸附CO,Co0可解離吸附,協(xié)同催化生成HA,同時Co和LaAlO3間存在電子轉化可提高活性。在260℃、3.0MPa、GHSV為900mL/(gcat·h)、H2/CO=2條件下,總醇選擇性為42.4%,HA選擇性為61.1%。Chen等通過原位DRIFTS、XAS、本征動力學等論證HAS活性位點位于Co-CoOx界面。Co和CeO2之間強烈的金屬-載體相互作用延緩了CoOx的還原并穩(wěn)定了Co-Coδ+中間體,見圖4,有利于CO在金屬Co表面解離形成CHx,在Coδ+位點上被締合活化。優(yōu)化單金屬Co催化劑催化性能,首先應通過形成Co-CoOx來供給足夠量的Co0和CoOx位點,因此需要兩種組分緊密結合。在實際反應條件下,Co-CoOx結構還會受活化和反應過程中溫度、壓力和氣氛等影響。
Co/CeO2催化劑上HAS過程示意圖
為解決Co單金屬催化劑HA時空收率低等瓶頸,可使用促進劑和/或載體調控電子和幾何性質,優(yōu)化吸附分子CO及烷基物種的比例,進一步提高催化劑性能。Lebarbier等結合原位表征及DFT計算發(fā)現,對于C—C耦合(包括CO插入)能壘,Co和La2O3之間的邊界比Co或Co2C位點更低。La摻雜增強Co2C相形成,進而提高Co2C/Co比。Co2C對CO插入所有CHx(x=1~3)活化能壘較低,如碳化物位于La2O3附近,則活化能壘更低,由此促進HA的形成,使HA選擇性隨Co2C/Co比率的增加而增加。減小Co顆粒尺寸或擴大La2O3與Co/Co2C間界面邊界效果更顯著。在225℃、3MPa、GHSV為500h-1條件下,15Co0.5La/AC轉化率為58%,醇選擇性為20.4%,總醇中HA選擇性達到88.8%。Liu等制備Co3O4-m介孔催化劑,通過TEM、原位XRD等表征發(fā)現有序介孔結構可促進碳鏈生長,生產更高碳數的醇和碳氫化合物。Co3O4-m及Pt/Co3O4-m催化劑HA總產率分別為1.6mmol/(gcat·h)、1.5mmol/(gcat·h)。
最新研究證實,在HAS反應中氧空位可與金屬Co、Co2C起到良好的協(xié)同作用。Song等通過檸檬酸鹽絡合法制備Ca1-xLaxTi1-xCoxO3催化劑,研究發(fā)現金屬Co NPs促進CO解離吸附和C-C偶聯(lián),并與吸附在Co2C上的*CO結合生成醇。氧空位可用于活化含氧化合物和促進合成HA。為引入大量氧空位,Guo等在鈣鈦礦的B位加入Fe,并通過原位還原和拓撲化學碳化制備氧空位增強的La0.9K0.1CoxFe1-xO3/ZrO2催化劑。原位CO-DRIFT未在Co2C上發(fā)現CO吸附,且醇選擇性與表面氧空位數呈線性相關,證實氧空位是生成HA的關鍵活性位點。程序升溫脫附(TPD)結果表明氧空位為CO提供了額外的吸附位并調節(jié)了CO的吸附形式,在Co、Co2C和氧空位的協(xié)同作用下,醇選擇性達到57.1%。
03
雙金屬CoCu催化劑
Co基催化劑研究多基于含Cu配方,然而Co、Cu表面能差異所帶來的相分離是亟待解決的難點。1978年法國研究所(IFP)發(fā)表的專利以CoCu為核心組分,認為高活性源于催化劑前體均勻度,助劑作用次之。為剖析CoCu催化劑構效關系、減少相分離、提升催化性能,最新研究重點在于通過表面修飾、添加助劑、優(yōu)化載體等手段調控催化劑構型、活性位點比例及分布等。
3.1 雙金屬作用
CoCu雙金屬催化劑具有協(xié)同效應、幾何效應或耦合效應,電子轉移利于催化反應,同時抗燒結抗積炭能力強。比單金屬催化劑具有更優(yōu)異的催化活性,且有望大幅度降低成本。但表面能和與吸附質結合強度差異大,CoCu合金易發(fā)生表面偏析。Liu等研究CoCu(111)表面偏析和CO覆蓋率變化對CO2加氫反應活性的影響,發(fā)現C—O鍵斷裂與*CH2O中間體進一步氫化之間的關系是生成乙醇的關鍵。適度Co偏析可提供合適的表面Co環(huán)境,具有共吸附*CO的橫向相互作用,從而提高乙醇選擇性。應用兩步還原誘導,在反應開始時將CoCu表面調節(jié)為中等Co偏析狀態(tài),乙醇選擇性達到60%以上,同時完全避免甲烷生成。
Co和Cu間的密切接觸是HAS的關鍵因素,Cu和Co的優(yōu)良混合提高了CO分子吸附在Cu上的概率,并提高了其遷移到吸附在Co上形成醇的CH中間體的可能性。優(yōu)選的CoCu基催化劑可同時實現高CO轉化率和HA選擇性。為研究CoCu雙位點作用,高娃等配制CuxCoy催化劑并調控雙活性位結構及CuCo間電子相互作用。結合結構表征與性能測試,發(fā)現通過優(yōu)化Cu對Co的分割效應、CuCo幾何與電子效應,使CuCo元素均勻高度分散,同時增強了雙活性位間的協(xié)同催化作用,從而實現催化活性、HA選擇性及穩(wěn)定性顯著提升。在220℃、2MPa、H2/CO=2、GHSV為2000mL/(gcat·h)條件下,CO轉化率為31.5%,總醇選擇性為50.6%,總醇中HA選擇性為80.8%。Liu等在Cu/Al2O3中加入Co,發(fā)現Co含量增加提高了煅燒樣品中CuCo2O4濃度。活化過程中CuCo2O4增強H原子吸附,促進形成分散良好的CoCu雙金屬位點,CO轉化率由7.6%提高到31%,HA選擇性從4.7%提高到31%。
3.2 分散性
提升CoCu活性金屬組分分散性是調控CoCu催化劑性能的重要策略。具體方法包括優(yōu)化制備方法控制金屬組分在載體上的分布和分散性、選擇合適金屬前體提高金屬組分的溶解性和分散性、添加助劑或表面活性劑改善金屬與載體間相容性和分散性、合成納米線或納米片特殊結構等。
采用微波輔助、超聲輔助等特殊合成方法,可在溫和條件下獲得分散均勻、尺寸小的金屬顆粒。Xu等采用輝光放電等離子體輔助浸漬法制備CoCu/Al2O3催化劑。在煅燒和活化之前,將浸漬態(tài)固體暴露于輝光放電等離子體(60Pa、13.56MHz、100mA和50mA柵極流)中,增加了比表面積并顯著增加了Cu分散。活性組分Co和Cu在表面富集,CO轉化率從27%提高到38%,HA/甲醇(摩爾比)從2.0提高到2.2。同技術應用在CoCu/SiO2催化劑上,發(fā)現焙燒前的等離子體處理有利于活性,但與HA選擇性幾乎無關。基于XPS和催化結果提出,Co物種的表面富集可以促進CHx的形成,從而支持碳鏈的生長。劉瑞琴等采用蒸發(fā)誘導自組裝法(EISA)制備CuCoZr催化劑,并以原位XRD、TEM、TPR、CO-TPD和原位DRIFT進行表征。發(fā)現EISA法制備的CuCoZr催化劑為有序介孔結構,Cu3Co1Zr催化劑Cu晶粒尺寸僅為9.1nm,CO轉化率達到74.9%,總醇中HA時空收率達到75.2mg/(gcat·h)。
前體直接關系到催化劑化學組成和結構,從而影響其催化性能,使用金屬鹽、配合物等作前體,可以提高金屬組分在載體上的溶解性和分散性。水滑石層板金屬呈原子水平高度分散、層間陰離子按一定方式有序排布且組成可調,近年來備受關注。寧珣等分別以CoZnAl-Cu(C2O4)2-LDHs和CoCuZnAl-CO3-LDHs為前體,經高溫拓撲轉變,制備插層結構及層狀結構CoCu-ZnAl2O4催化劑。發(fā)現比起傳統(tǒng)浸漬法制備的CoCu/ZnAl2O4催化劑,LDHs為前體使CoCu雙金屬中心具有更加均勻的分散結構,在HAS反應中表現出優(yōu)異的活性和HA選擇性。高娃等選取不同Cu/Co比的CuCoAl-LDHs為前體,系統(tǒng)調變LDHs前體的焙燒/還原條件,并采用晶體原位生長技術,制備得到核殼結構負載型Cu@(CuCo-合金)/Al2O3催化劑。該催化劑HAS催化活性優(yōu)異而且HA選擇性顯著提升,C6+醇選擇性達到48.9%,同時穩(wěn)定性強。SUN等在LDHs前體中同時引入Cu、Co元素,使CuCo顆粒小且分散均勻,顯著提高催化活性和醇選擇性,C1~C3混合醇達到90.6%。Cu和Co物種的良好混合提高了CO以橋接幾何結構結合到相鄰金屬Co原子的概率,有利于CO的解離,促使CHx中間體增加。程序升溫表面反應(TPSR)證實CuCo均勻分散,CHx加氫生成CH4概率低,CHx中間體更傾向于生成乙醇。
制備納米線或納米片等具有納米尺寸的催化劑顆粒,可以調節(jié)表面能,有效促進金屬組分的分散。為改變表面Cu+/Cu0分布和Co的分散性,Sun等制備獨特“球片狀”結構的CuCoMn NPs催化劑及CuCoAl納米片催化劑。CuCoMn NPs催化劑經簡單共沉淀再煅燒-還原得到富CuMn的球狀結構和以Co為主的納米片。通過HAADF-STEM、原位XRD和原位DRIFT等表征發(fā)現Cu和Mn間電子和幾何相互作用較強,改變了Cu的化學狀態(tài),使表面Cu+/(Cu0+Cu+)比例增加。尤其增強了Cu+活性位點上的CO線性吸附,使CO插入概率更高。HAS產物中總醇選擇性為46.2%,總醇中乙醇達45.4%。通過共沉淀法合成CuCoAl納米片催化劑,Al顯著增強Cu和Co的分散,納米片上豐富的堿性位點(表面羥基組)使甲酸形成,甲酸是生成CHx中間體的重要C1分子。在Co原子上吸附甲酸和碳橋產生了更多CHx中間體,利于C—C鏈生長。HA選擇性達到54.9%,乙醇/HA比例達到55.9%。
選擇適當載體、構建特殊催化劑結構,可以使Co和Cu與載體或其他材料產生較強的相互作用,改變Co和Cu在不同層次或區(qū)域分布,穩(wěn)定金屬組分并減少相分離。Li等將催化劑封裝在KIT-6介孔硅分子篩內部,原位生長合成CoCu-Z@KIT-6催化劑。KIT-6的逐步熱解和限制促進了CoCu高度分散,使其具有分散良好的金屬顆粒和豐富的CoCu合金位點,有利于CO橋式吸附位點具有較高的CO解離能力。促進了CO解離與CO插入位點之間的協(xié)同催化,催化性能提高,C2+醇STY為31.9mmol/(gcat·h)。高度分散和原子緊密相互作用的CoCu雙金屬位點的存在使催化劑具有良好的HAS穩(wěn)定性。Dokuchits等采用檸檬酸鹽法合成高分散LaCo1-x-yCuxTiyO3/KIT-6鈣鈦礦,并溶解在7% NaOH水溶液中去除KIT-6基質。結合表征手段認為醇的合成發(fā)生在3nm雙金屬CoCuNPs和4~6nm的Co顆粒上。在240℃、2MPa、GHSV為900h-1條件下,CO2選擇性低至5%,C2~6醇選擇性為43.5%、STY為21mg/(gcat·h)。
添加適當的表面活性劑或穩(wěn)定劑可以改善金屬組分與載體之間的相互作用,減少金屬顆粒的團聚,提高分散性和抗燒結的結構穩(wěn)定性。Jiao等基于TPR-MS、TPSR-MS等認為適量La增加了Co的分散性,降低了催化劑還原性,促進反應中Co2C生成,調節(jié)了Con+/Co0比例。將質量分數0~2%的La與質量分數15%的Co共浸漬在AC上,CO轉化率和總醇選擇性隨助劑負載量呈火山型變化,La質量分數0.5%時,最大值分別為21%和39%,并且所有樣品甲醇選擇性均低于4%。Zhu等發(fā)現La改性CuCo基催化劑形成的納米片結構產生了更多氧空位。LaxCuCo催化劑解離和締合CO吸附能力顯著提高。表面活性位點(Cu0、Co0和氧空位)的適當協(xié)同作用使催化劑表面更易形成甲酸鹽和甲氧基中間體。加入La降低了納米片厚度,并作為隔離島抑制晶粒聚集和生長,使反應中LaxCuCo保持高分散。La0.3CuCo催化劑CO轉化率為25.4%、總醇選擇性為61.4%、總醇中HA選擇性為79.03%,并且穩(wěn)定性優(yōu)異。
對載體進行預處理,如官能團修飾、表面氧化或還原等,可增加載體與金屬組分之間的相互作用,提高金屬分散性。Zhao等研究發(fā)現MnAl載體避免了活性相團聚,高溫還原后形成分散均勻的CuCo合金顆粒。雖然干凝膠前體煅燒后不可避免地形成了一些CuO和Co3O4,但CuCo/MnAl氧化物前體中的Cu和Co物種高度均勻地分散,尺寸小,催化劑主相是CuCo2O4。催化劑還原后的TEM和HRTEM圖見圖5(a)、(b),CuCo顆粒高度分散且尺寸分布集中在5~8nm,圖5(c)顯示兩種元素相伴共存,且在載體上均勻分散。
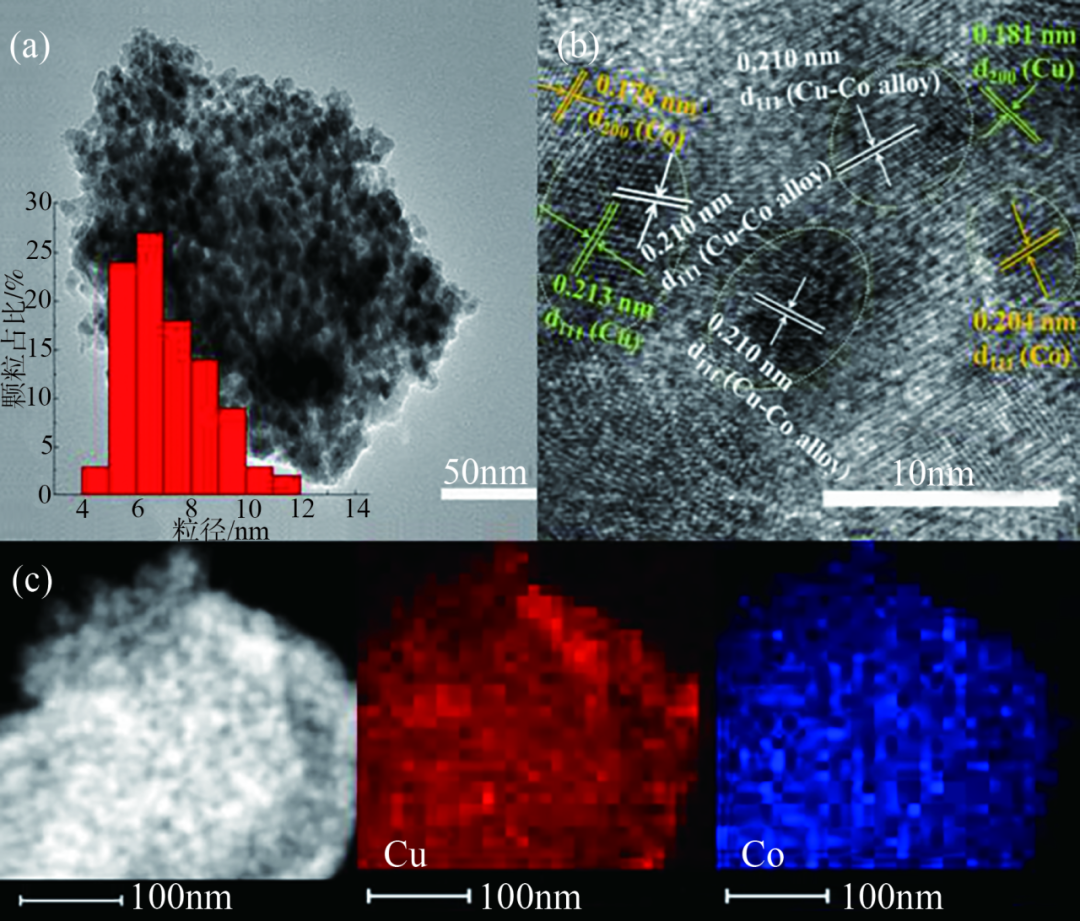
還原后3Cu5Co/MnAl催化劑的TEM和HRTEM圖像
3.3 金屬比例
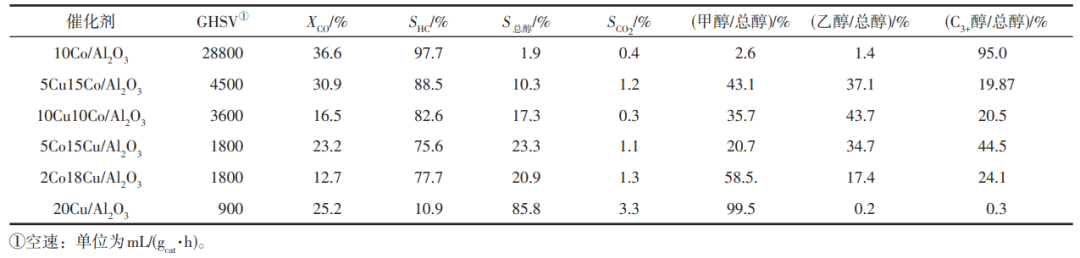
Cu/Co比例調控對CoCu基催化劑性能起決定作用。Wang等以共初濕浸漬法制備不同Co/Cu質量比材料,結合原位XRD、XPS、TPR、XANES/EXAFS、原位磁法和TEM等多種表征技術,認為Cu在γ-Al2O3上的分散度遠高于Co。在負載的Co催化劑中添加少量Cu后,CO加氫的速率和選擇性都發(fā)生了顯著變化,Cu的存在使醇的選擇性增加了一個數量級,并降低CO的總轉化率。不同金屬比例催化劑在250℃、2MPa、H2/CO=2條件下性能對比見表1。Su等制備不同Co/Cu比納米粒子催化劑進行動力學研究,調控催化劑金屬比例,得到的催化性能趨勢相同。CO加氫主要發(fā)生在Co位點,加入Cu提高了HA選擇性,但Cu原子覆蓋了部分Co位點使活性降低,同時也降低了CH4選擇性。在Co5Cu2.5/SiO2催化劑上總壓力5MPa、不同溫度下研究反應的本征動力學,發(fā)現所有與CO相關的反應級數都是負的,與H2相關的為正,形成醇的活化能約為形成烴的一半。
表1 不同金屬比例催化劑性能
結合DFT、微觀動力學,Prieto等研究Co/Cu(摩爾比)為3/0、2/1、1/2、0/3的CoCu NPs催化劑表面上產物選擇性,Cu、Co表面位點比例對CH4、甲醇、乙醇選擇性的影響。發(fā)現最佳Cu/(Cu+Co)(摩爾比)為0.33,最高總醇時間產率為27mmol/(gCu+Co·h),富Co表面的合金化CoCu NPs更利于反應。CuCo雙金屬體系復雜,Co、Cu和Con+共存,而后兩者功能相似。因此,如何在CO加氫過程中平衡Co和Cu的含量、控制Co的電子狀態(tài),是進一步研究的挑戰(zhàn)。Zhao等采用溶膠-凝膠合成法制備不同Cu/Co(摩爾比)的MnAl氧化物負載CuCo合金催化劑,并利用XRD、BET、TEM、XPS、H2-TPD/TPR、CO-TPD、原位CO吸附DRIFTS、CO-TPSR等進行表征。通過優(yōu)化Cu/Co比例調節(jié)催化劑表面對CO加氫和插入的吸附氫、非解離吸附CO的量,提高還原性,改善HAS催化行為。隨Cu含量增加,CO非解離吸附量相應增加,3Cu-5Co/MnAl催化劑催化性能最佳,在5000h-1、260℃和5MPa條件下CO轉化率為33.4%,總醇選擇性為39.7%,總醇中C2+醇選擇性為57.3%,時空產率達到63.0mg/(mL·h)。此外,CO2選擇性隨Cu/Co摩爾比提高而增加,證明金屬Cu有利于WGS反應。
04
助劑調控催化性能
在催化劑體系中增加助劑組分進行調控是催化劑性能優(yōu)化的重要手段,Co基催化劑體系助劑的研究重點為堿金屬(AM)、La2O3、Rh等。三元體系中CuCoMn表現顯著,優(yōu)于CuCoCe、CuCoNb和CuCoMo。
4.1 AM改性
AM被廣泛用于調節(jié)催化劑表面形貌、晶體結構和孔隙結構,用其改性可促進碳鏈增長、提高催化劑活性和選擇性。羅文昭等為Co0.5Cu/γ-Al2O3催化劑添加各種AM助劑,發(fā)現除Cs外所有金屬助劑均提升了HA在總醇中占比,K使催化劑活性和產物HA選擇性同時提升,HA在總醇中的占比達到68.69%。Tien-Thao等比較不同AM助劑對納米LaCoCuOx鈣鈦礦催化劑HAS反應的影響,認為AM使混合氧化物比表面積增加,并遵循Li<Na(~Cs)<K<Rb趨勢,同時總醇生成率提高。AM改善了碳鏈生長、抑制了甲烷化,但同時促進了WGS活性。Cosultchi等以2%(質量分數)AM(Li、Na、K、Rb和Cs)對混合CoCuCr氧化物進行改性。加入Na使HA選擇性達到34%,略高于HC選擇性(33%),但CO2選擇性上升到30%。為對比Na和K促進作用,Ao等以La0.9Sr0.1Co0.8Ni0.1Cu0.1O3鈣鈦礦前體為原料制備三金屬CoNiCu催化劑。發(fā)現325℃下K促進的催化劑醇分布為82.2%,醇鏈生長概率因子為0.53,優(yōu)于Na促進和非促進催化劑。添加堿性助劑一方面促進穩(wěn)定的Co2C形成、提高Co2C/Co比,醇選擇性增加但同時降低了CO轉化率;另一方面提升堿度促進了醇的碳鏈增長,尤其對甲醇轉化為乙醇。因堿性助劑增加催化劑堿性,反應中促進吸附和活化,防止了CoNiCu催化劑碳沉積失活,特別是K在340℃下仍可保持催化劑性能穩(wěn)定
4.2 Zr/Zn改性
Ge等研究發(fā)現ZrO2在CoCu、CoFe催化劑上呈無定形且有缺陷,可增強H2和CO活化,促進金屬和碳化物相形成。ZrO2在5%~10%(摩爾分數)(Cu∶Co=1∶4,Co∶Fe=1∶4)性能最佳,HA生產率可分別提高2~5倍,且300h內無失活跡象。Co1Fe4@ZrO2-10的HA選擇性最高為31.8%、STY最高為345mg/(h·gcat)。郭磊等考察助劑Zn、Zr、La和Mn的影響,發(fā)現對CO解離速率、Co2C生成及HA選擇性促進作用明顯,構筑的Co2C/Co0構成雙活性位結構。Zn改性Co/AC催化劑CO解離速率、活性和HA時空收率相對最高。Huang等以CuCoAl和ZnO/ZrO2復合材料制備CuCoAl|ZnO/ZrO2改性催化劑,如圖6所示。當ZnO/ZrO2(摩爾比)為4∶1時,總醇選擇性最高達到42.6%(質量分數),HA占總醇83.7%。結合各種表征發(fā)現因缺乏CO*或CH*,應控制H*/C*比率在一定范圍才可生成HA。ZnO/ZrO2=4∶1的H*/C*比例適度,保持了CO*、CHx*和H*相對量的良好平衡,促進了CnHx*-CO*偶聯(lián)反應,從而提高總醇及HA選擇性。
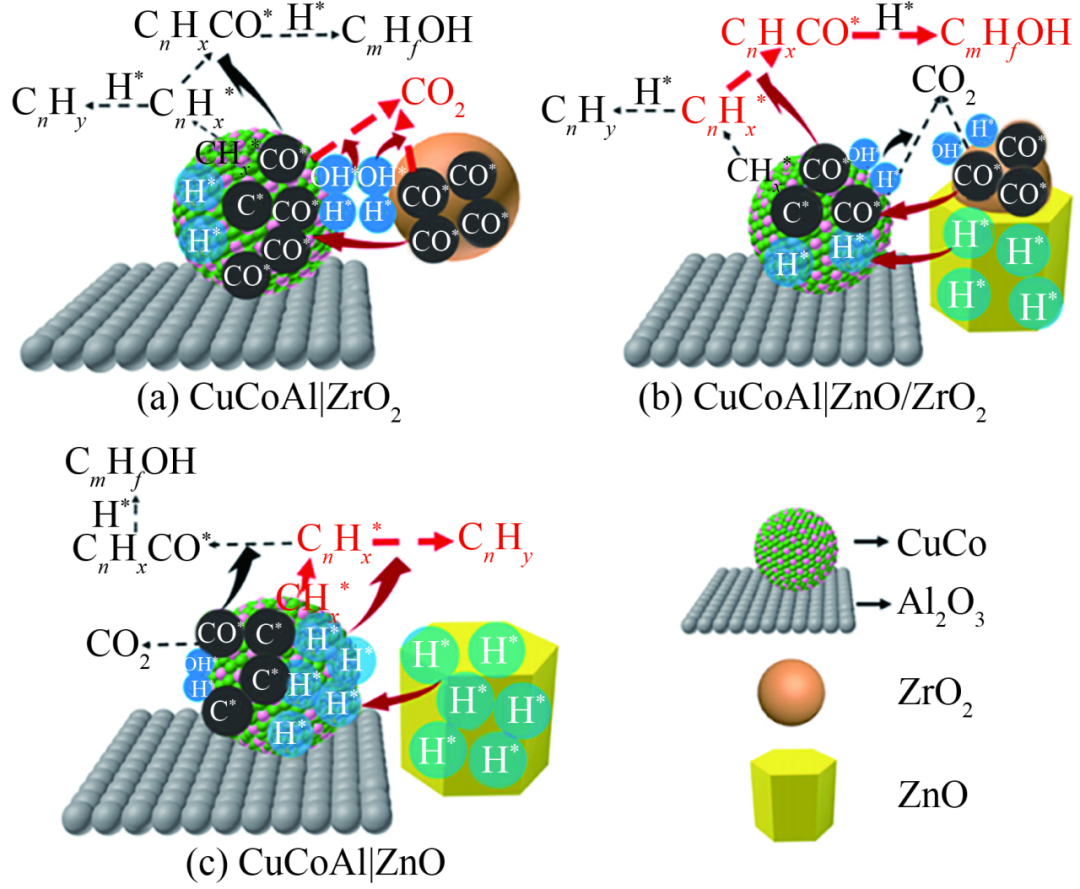
催化劑HAS過程示意圖
4.3 第Ⅷ族改性
Rh的典型作用是利于乙醇生成,也可作為Co基催化劑助劑。Li等發(fā)現1% Rh(質量分數)可明顯改善CoCu/Al2O3性能,原位漫反射紅外和H2-TPR證實了Rh優(yōu)越的氫化能力,使CO轉化率從27%提高到37%,總醇選擇性從16%提高到34%,HA/甲醇比從1.4提高到1.7。Qin等將Rh或Ru組分摻雜到CoMn催化體系中極大促進了HAS活性和HA選擇性。1.1Rh-CoMn和0.7Ru-CoMn的活性比CoMn催化劑提高2.5倍以上,高碳數含氧化合物的生成率分別提高4.1倍和2.7倍。高度分散的Rhδ+或Ruδ+位點提供了大量的表面碳相關物種,并創(chuàng)造了局部富C化學環(huán)境,促進了Co2C形成和穩(wěn)定,同時增加了非解離CO*物種的表面覆蓋率。利于構建HAS的Co2C-Co雙位點及生成含氧化合物,但Rh的高價格限制著此類研究的興趣。
最新的研究還包括CoFe合金等,Zeng等提出了一種由CoxFe3-xO4尖晶石氧化物NPs衍生的單分散ε′-(CoxFe1-x)2.2C合金碳化物催化劑并應用于HAS反應。Co/Fe(摩爾比)對活性位點的演變影響顯著,Co/Fe(摩爾比)為1/2時催化劑ε′-(CoxFe1-x)2.2C含量最高達到80.2%,HA選擇性和時空產率最高,分別為38.5%和1.932g/(g(Fe+Co)·h),HAS性能超越大多數已報道的改性FTS催化劑。DFT計算證實當Co/Fe比超過1/2時,形成ε′-(CoxFe1-x)2.2C合金碳化物變得更加困難。ε′-(CoxFe1-x)2.2C與CO適度的鍵合,對CO解離和CO插入之間的平衡至關重要,可提高HA選擇性。由于抑制了相分離和表面碳沉積,具有堅固CoFe合金碳化物結構的Co1Fe2催化劑性能穩(wěn)定超300h。
4.4 Ga改性
為研究Ga對CO插入和C—C耦合的Co位點的促進作用,An等制備顆粒分散均勻的負載型CoGa催化劑,發(fā)現與Co原子相鄰的Ga原子有助于隔離Co中心,并向相鄰Co位點提供電子。高度分散的Co位點負責線性非解離CO吸附,促進CO插入步驟以生成醇。GaCo位點的電子供給促進了游離CO吸附,從而促進碳鏈生長。CO插入和C—C偶聯(lián)的匹配,增強了HAS活性并提高了HA選擇性。Gao等以Ga摻雜Co基催化劑,發(fā)現有利于提高醇選擇性。優(yōu)化后的15Co2.5Ga/AC催化劑醇選擇性達到30.3%(摩爾分數),無Ga催化劑僅為17.0%。Ga摻雜提高了Co3O4還原溫度,改變了表面Co2+和Co0物種分布,適度比例的Co2+/(Co2++Co0)使醇選擇性增強,添加Ga是提高HA選擇性的有效途徑。
郭海等通過實驗驗證CoGa合金中Ga0對CoGa顆粒的穩(wěn)定性具有促進作用。通過調變金屬比例,發(fā)現Ga/Co比為0.2~0.5時形成具有阱結構的CoGa顆粒,Ga/Co為比大于1.0不再具有阱結構。Ga/Co比為2.4和8.0時不具有阱結構,但表面存在大量Ga0,隨Ga0/Go0比增加,C2+醇選擇性增加,烷烴選擇性降低,穩(wěn)定性好。An等以Co—Al—O尖晶石前體合成Ga摻雜Co基催化劑,在270℃、3.0MPa條件下,CoGa1Al1O4/SiO2催化劑乙醇選擇性達到20.1%。Ga和Co間的強相互作用誘導Co0-Coδ+活性對構型,通過偶聯(lián)解離(CHx*)/非離解(CO*)中間體促進乙醇合成,為CO和CO2氫化的HAS提供優(yōu)勢途徑。
4.5 其他改性
在CoCu催化劑中摻雜La元素抑或以檸檬酸鹽絡合法制備鈣鈦礦型氧化物材料,均可使催化劑中的混合金屬高度分散,進而調控催化劑性能,典型催化劑為CoCuLa2O3、CoCu/LaFeO3、CuO/LaCoO3、LaCo1-xCuxO3/ZrO2等。Wang等分別以La、Zr、Al摻雜CoCu催化劑,在3MPa、250℃、H2/CO=2、GHSV為36000mL/(gcat·h)條件下評價對比三種催化劑,發(fā)現CoCuLa2O3的C1~6醇選擇性為34.9%,遠高于CuCoAl2O3(13.1%)、CuCoZrO2(17.8%)。并且CoCuLa2O3催化劑HA選擇性最大,乙醇選擇性為10.5%,La2O3增加了表面堿度,有利于醇類形成。
Al、Mg等其他金屬改性也有研究。Li等以表面-固體反應構建Al和La改性的新型催化劑,通過Co和Al相互作用形成CoAl2O4活性結構,有利于保持催化劑中Co2+穩(wěn)定性,促進中間體CHx*和CO*偶聯(lián)。助劑La和Al可均勻分散在催化劑表面,并抑制活性位點燒結,轉化率是純Co催化劑的3倍,270℃時總醇選擇性達到39.1%。Pei等使用3.8%Al2O3和6.3%SiO2作Co[15%(質量分數)]/AC的促進劑。含Al2O3樣品CO轉化率為69%、總醇選擇性為28%,且醇中HA達到96%。Al2O3誘導了Co2C形成并改變吸附H2與CO的比例。含Si材料在總醇選擇性和助劑含量方面有優(yōu)勢,但催化劑活性僅為38%。SiO2的關鍵促進作用是抑制反應過程中Co聚集,并保留大量有利于CO插入的Co2+。
05
載體
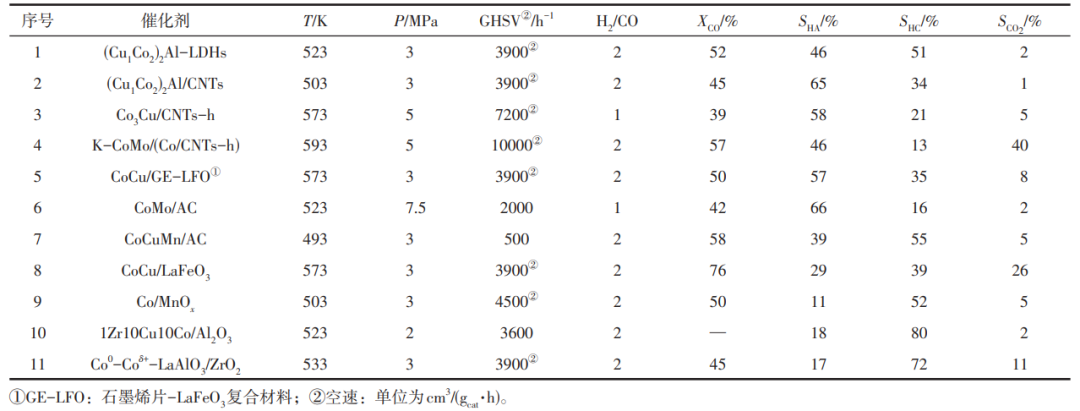
粉體催化劑中CuCo活性組分難于均勻分散,CuCo合金化程度低且協(xié)同作用弱,容易燒結、脫合金導致催化劑嚴重失活,優(yōu)化載體可大幅提高催化劑穩(wěn)定性。載體研究包含ZnO、SiO2、Al2O3、MgO、MMO、ZrO2、La2O3、AC、碳納米管(CNTs)、OMC和碳纖維(CF)等眾多材料,如表2所示。
表2 典型Co基催化劑載體性能
在相同CO轉化率水平對比Al2O3、SiO2和CNTs載體,發(fā)現以Al2O3為載體的催化劑總醇選擇性最高。載體上形成的CuCo雙金屬相在能量過濾TEM圖像中均勻分布且顆粒尺寸小。Lee等對比Al2O3、MgO和ZnO載體,通過NH3-TPD、SEM和TEM等表征發(fā)現Zn負載型催化劑轉化率相對更高(18%),歸因于其低配位氧原子及更高的酸性位點強度,具有HAS潛力,但甲醇選擇性高達83%。高娃等在Al基底上原位生長CuCoAl水滑石納米片,基于其結構拓撲效應得到結構穩(wěn)定的Cu@CuCo合金/Al2O3催化劑,見圖7,NPs核殼結構平均直徑15nm,載體的片狀形貌和開放通道利于質量傳輸并抑制熱點形成。C6+醇選擇性達到48.9%,80h性能穩(wěn)定。

Cu@CuCo合金/Al2O3催化劑SEM、TEM、HAADF-STEM圖像
LDHs具備的獨特層狀結構是催化劑載體改性的最優(yōu)方案之一。Liao等以LDHs前體為原料制備CoMn催化劑,發(fā)現高反應溫度下層狀結構比非層狀結構活性和醇收率更高。因層狀結構表面積更大,可避免Co、Mn分離和防止聚集,使催化劑顆粒尺寸更小、Co-Mn間相互作用更強,且活性位點在層上高度分散,穩(wěn)定性好。Cao等通過煅燒LDHs制備CoCu NPs,優(yōu)化后催化劑比表面積達106m2/g,晶粒尺寸僅4.8nm,CO轉化率達到52%。EDX圖譜和EDS線掃描圖譜表明金屬混合高度均勻,利于HA形成,總醇選擇性為46%,總醇中HA高達98%,同時CO2選擇性低至2.4%,見表2第1行。在200h催化性能穩(wěn)定,CoCu合金NPs僅略微增大至6.2nm。Wang等基于CF優(yōu)異的導熱性和LDHs中金屬離子混合均勻,以共沉淀法制備LDHs和CF納米復合物利于金屬合金形成。通過FTIR、XRD、SEM和TEM等技術表征證實,復合材料形成高度開放的多孔結構。還原后CuCo合金NPs催化劑表現出高活性和HA選擇性。
CNTs可抑制體相CuCo催化劑上HC形成,同時保持高活性和低CO2和甲醇選擇性,是近年來研究熱點。Shi等先以HNO3水溶液處理CNTs增加其孔隙率,提高金屬分布,再以酸改性。CO轉化率由15%提高到18%,總醇收率提高到150g/(kgcat·h)。Feng等以CNTs吸附在硅膠上制成CNTs-SG復合物載體,CO轉化率37%,總醇中HA選擇性62%(質量分數)。在CNTs-SG中引入鄰苯三酚使金屬分布和相互作用更佳,CO轉化率和總醇中HA選擇性分別提高到40%和73%(質量分數)。為解決反應放熱形成熱點導致的HA選擇性受限,Cao等在CNTs和CF上共沉淀和老化CuCo-Al LDHs,如圖8所示,將酸處理的CNTs分散在含有Cu2+、Co2+和Al3+陽離子的水溶液中。加入NaOH和Na2CO3作為沉淀劑,得到的懸浮液老化、洗滌、干燥并在723K下還原。(-)和(+)電荷分別表示酸處理后CNTs的官能團和添加的陽離子。CNTs催化劑CO轉化率為45%,總醇選擇性為65%,且其中97%為HA,見表2第2行。
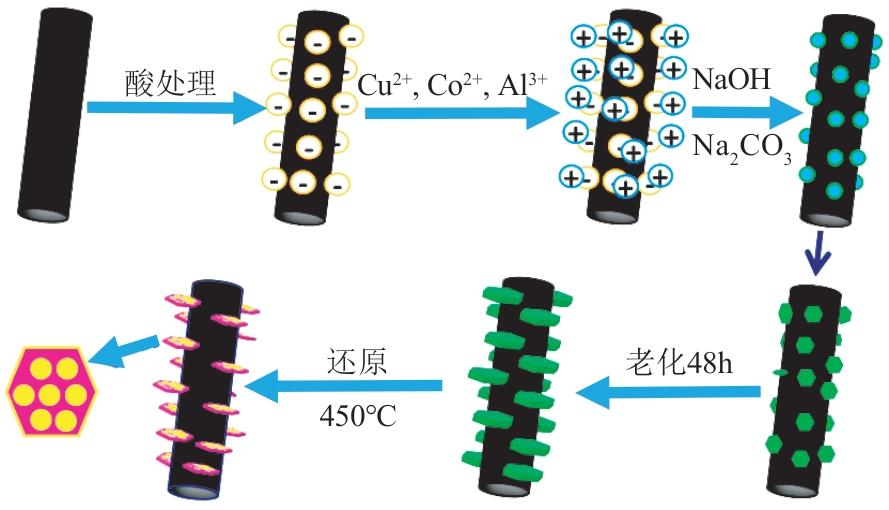
(Cu1Co2)2Al/CNTs制備示意圖
CHEN等合成疏水性和親氣性的多功能CuCoSNTs-c催化劑,見圖9,可抑制C1分子形成,提高CO轉化率和HA選擇性。中孔壁充分暴露了活性位點,管狀通道改善了傳質動力學,加速了醇產物的解吸。疏水性和親氣性管壁可調節(jié)CuCoSNTs-c局部微環(huán)境,富集CuCo活性位點周圍的H2和CO分子,并快速解吸HAS中原位產生的水分子。實現CO轉化率高達80.4%,HA選擇性高達66.6%,同時將CO2選擇性抑制在低于1.2%,360h內性能穩(wěn)定。
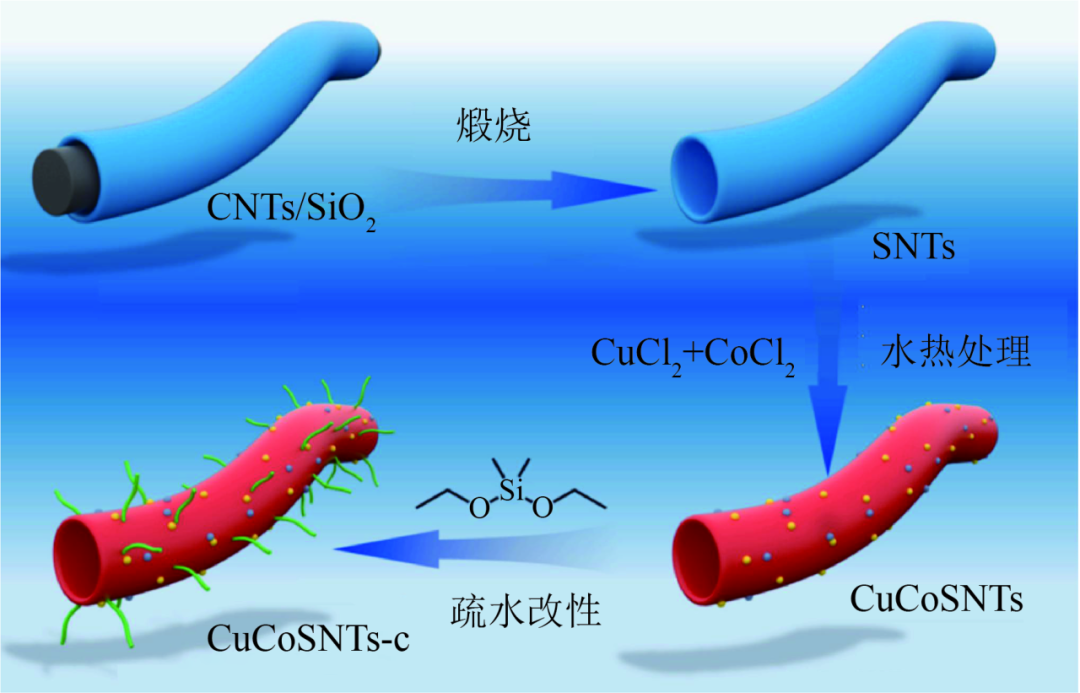
疏水性催化劑CuCoSNTs-c的合成示意圖
Karim等研究AC作支撐材料,CO轉化率為42%,CO2選擇性最小為1.7%,HA選擇性最大為66%,見表2第6行。Pei等利用初濕浸漬法將CoCuMn負載在AC上,基于HRTEM提出小Co NPs的大Cu簇可提高HA選擇性,CO轉化率為58%時HA選擇性為39%(表2,第7行)。Niu等制備一系列含石墨烯的復合材料負載CoCu NPs,石墨烯質量分數5%時性能達到最優(yōu)水平,見表2第5行,CO轉化率為50%,CO2選擇性低于8%,總醇選擇性為57%,HA占比達90%,運行160h未見明顯失活。Guo等通過熱合成層狀結構金屬有機框架(MOF)材料制備CoMn催化劑,N2氣氛下350℃煅燒后可保持部分MOF骨架。CO轉化率為51.5%時,醇選擇性為57.5%,總醇中HA選擇性為82.4%,250h穩(wěn)定性良好。Cui等發(fā)現核殼Co/MnOx@quasi-MOF-74催化劑使Co0、Co2C和Co2+配位不飽和位點間的協(xié)同作用增強,促進HA形成。quasi-MOF-74框架內Co0 NPs促進CO解離和CHx-CHy耦合,均勻分布的Co2+配位不飽和位點與Co2C協(xié)同增強CO插入。總醇中HA選擇性高達93.2%,同時CH4、CO2選擇性極低。
06
結語
HA在化學和能源工業(yè)中應用廣泛,生產HA的糖發(fā)酵和烯烴水合法分別存在可擴展性有限和單程轉化率低的問題。HAS可從非常規(guī)天然氣和可再生能源中獲取原料,在替代路線中尤具吸引力,但催化劑活性和HA選擇性離成功商業(yè)化仍存在差距。Co基改性FTS催化劑在過去十多年中受到分子水平研究的高度關注,有巨大改進,是目前最有前途的材料。
(1)結合FTS和MS領域最新見解分析HAS機理,包括從CO活化到形成HA的最佳路徑等。論述Co0和Con+位點作用、Co和Co2C相間協(xié)同效應、CoCu結構域及CoCu(100)等表面在HAS中的作用。
(2)Co0-Coδ+單金屬催化劑HA時空收率低,可優(yōu)選促進劑和/或載體調控電子和幾何性質,調控吸附分子CO及烷基的比例進行改善。
(3)CoCu合金的協(xié)同作用使催化劑具有高活性和高選擇性,顯著抑制所有C1分子產生,減緩HC形成。Co、Cu表面能差異所帶來的相分離是亟待解決的難點,可通過表面修飾改變表面能、提高CoCu合金化程度、合成特殊結構、摻雜助劑或優(yōu)化載體等方法進一步提高性能。
(4)助劑K改善碳鏈生長、抑制甲烷化、防止碳沉積、耐高溫穩(wěn)定性優(yōu)勢明顯。Zr/Zn促進CO解離、Co2C生成,提高HA選擇性。Rh/Ru極大促進HAS活性和HA選擇性。Ga改變表面Co2+和Co0分布,提高醇選擇性。
(5)載體研究重在具備獨特層狀結構的LDHs,及CNTs、AC等眾多碳基材料。CNTs可抑制催化劑上HC形成,同時保持高活性、低甲醇及CO2選擇性。
(6)未來研究方向為對機理的深度剖析、活性位點交互作用、新型載體材料開發(fā)等,在高轉化率基礎上,進一步提高HA選擇性,減少HC選擇性。
作者簡介

第一作者:張琪,碩士,高級工程師,研究方向為煤間接液化。
通信作者:門卓武,博士,教授級高級工程師,研究方向為煤間接液化;呂毅軍,博士,高級工程師,研究方向為煤間接液化。


